
La Directiva ICH Q3D tiene como finalidad última limitar la presencia de impurezas elementales (o también llamados metales pesados) potencialmente tóxicas en los productos farmacéuticos destinados al ser humano.
Esta directiva va unida a cambios en las Farmacopeas (EP & USP) con la introducción de nuevos métodos analíticos más selectivos, más precisos, con mejor recuperación y por otro la directiva justifica los limites con relación a la toxicidad de los elementos potencialmente presentes.
La directiva fija una lista de 24 elementos repartidos en cuatro categorías, con arreglo a su toxicidad y con arreglo a su probabilidad de ocurrencia y la dosis diaria máxima admisible (PDE: Permitted Daily Exposure) para cada impureza según la vía de administración (µg / día).
La directiva fija la metodología para:
- Los elementos que hay que tener en cuenta.
- Los límites para cada elemento.
- La metodología a seguir en la realización de los análisis de riesgo.
- Las estrategias de control en función de los valores/limites obtenidos.
Las fechas de implementación de esta nueva normativa son:
- Nuevos registros: 1 de junio de 2016.
- Productos ya comercializados: 1 de enero de 2018.
¿CÓMO EMPIEZO A IMPLEMENTAR LA NUEVA DIRECTIVA?
Hay que tener en cuenta 3 temas antes de empezar el análisis de riesgos:
- Las estrategias que sigamos va a depender en gran parte de la información de partida de los proveedores
- Lo normal es que el laboratorio farmacéutico no disponga de los nuevos métodos analíticos para la determinación de las impurezas elementales.
- El plan analítico no se puede plantear hasta no haber analizado toda la información disponible de partida.
El proceso de realización del análisis de riesgos a nivel práctico es el siguiente
- Carnet de identidad del medicamento
En esta etapa hay que recoger toda la información disponible del medicamento: formula unitaria, especificaciones, datos analíticos disponibles, datos de proveedores, , proceso de fabricación, servicios,…..
- Elegir el enfoque
2 potenciales enfoques:
- El enfoque del componente: El análisis de riesgo está basado en los niveles de impurezas elementales presentes en cada componente (API, excipientes, proceso de fabricación, servicios y material de acondicionado) que puede aportar IE al medicamento
- El enfoque del medicamento: El análisis de riesgo está basado en los niveles presentes en cada medicamento mas la posible interacción con el material de acondicionado.
- Recoger los datos
En la Estrategia del medicamento los datos derivan de:
- Análisis del medicamento.
- Interacción con el material de acondicionamiento.
- Necesario lotes representativos que cubra todas las fuentes de variabilidad. (proveedores, equipos, orígenes, excipientes,…)
En la estrategia de los componentes los datos provienen de cada uno de los componentes:
- Informaciones proveedores (cuestionario, COA, certificaciones, …).
- Bibliográficos.
- Datos generados sobre equipos y servicicios
- Interacción con el material de acondicionamiento.
- Análisis de los componentes
- Evaluación del riesgo
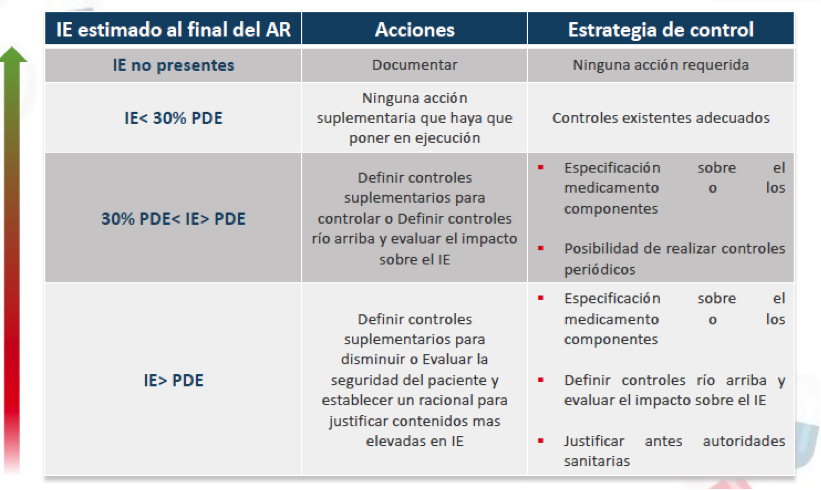
Para ello se compara el valor obtenido con respecto al valor de PDE de cada una las impurezas elementales. En esta etapa se debe definir el valor de concentración máxima a partir del valor de PDE según las distintas estrategia de conversión (1,2A, 2B y 3)
- Estrategia de control
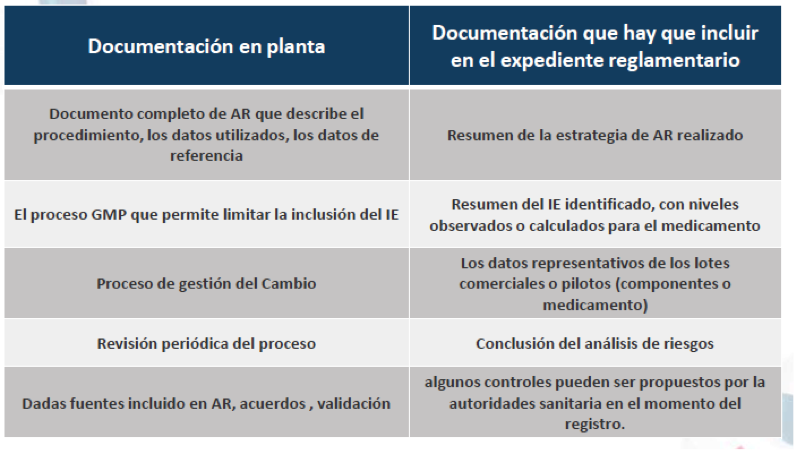
 6. Documentación
6. Documentación
- Ciclo de vida
El control de cambio que recoja cualquier cambio en el ciclo de vida de un medicamento (cambio de proveedores, cambios de proceso, material de acondicionado, excipientes,….) debe tener en cuenta el impacto sobre los niveles de impurezas elementales que puede originar dicho cambio.
Implementación ICH Q3D (Impurezas elementales)
Nuevos métodos analíticos más selectivos, más precisos, con mejor recuperación que minimizar el impacto económico, para el análisis de riesgos de implantación de la ICH Q3D

1 comentarios
Odette Piffaut
Muy bueno el resumen!!
Gracias