
En este tercer artículo de la serie basada en Anexo 1, vamos a analizar los cambios que se han llevado a cabo en operativa de planta, en aquello referido a personal y monitorización.
Empezaremos por el personal, ya que como sabemos, “La correcta fabricación de medicamentos depende de las personas.”
El concepto básico en cuanto a personal sigue vigente: debe tener experiencia, formación y actitud adecuadas, con el foco puesto en la protección del producto estéril en todas las fases del proceso. Es responsabilidad del fabricante asegurar que se dispone de suficiente personal y que este sea apropiado y esté debidamente cualificado y experimentado.
Se da en este borrador una especial importancia a la formación, especificándose los ámbitos en que el personal relevante debe estar formado. Se asume que dicha formación debe ser completa, siendo acorde a la criticidad de las actividades y áreas de trabajo a desarrollar por cada uno.
Formación integral del personal que trabaja en áreas estériles
Se restringe la entrada del personal no cualificado a Grados A y B, especificando que para los casos excepcionales en que sea necesario su acceso, existirán procedimientos escritos en que se describa en qué situaciones estará permitido y los pasos a seguir; se debe incluir la supervisión por una persona cualificada y autorizada. Además, será necesario evaluar el impacto de dicho acceso sobre la limpieza de las áreas.
Recordemos que además de un procedimiento de cualificación del personal, debe haber un sistema de descualificación, en el que se definan los motivos de descualificación (como la participación en Aseptic Process Simulation (APS) fallido o tendencias negativas en la monitorización de personal). Para permitir que un empleado descualificado vuelva a participar en prácticas asépticas, debe haber un reentrenamiento y una recualificación. Para que pueda acceder a Grado B y realizar intervenciones en Grado A, un empleado que ha sido descualificado debe, además, participar en un APS con resultado conforme.
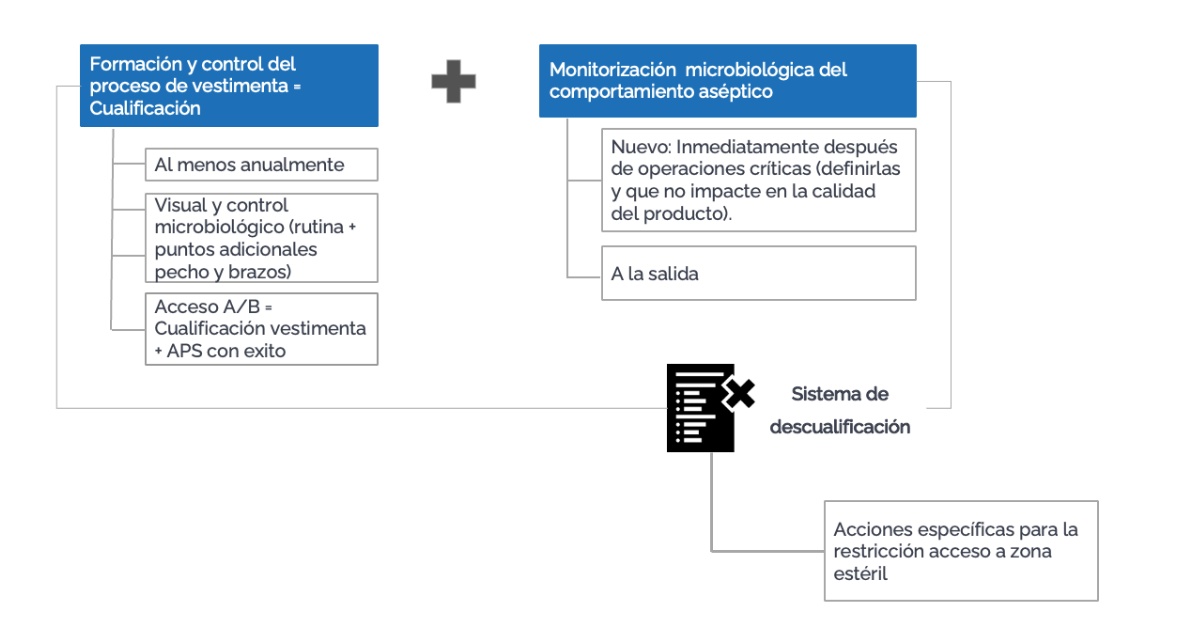
Muy relacionado con el personal y para garantizar la calidad del aire de las salas, tendremos el programa de monitorización, que estará ahora conformado por tres pilares:
- Monitorización ambiental – partículas no viables
- Monitorización ambiental y de personal – partículas viables
- Simulación del proceso aséptico (solo para productos de fabricación aséptica)
La monitorización ambiental, como todas las estrategias para controlar la contaminación del producto en esta nueva versión del Anexo 1, debe estar basada en un Análisis de Riesgos que nos dé soporte para definir frecuencia, localización y condiciones de incubación de las muestras. Así, consideraremos para la realización de este Análisis de Riesgos todos aquellos elementos que puedan afectar a la contaminación de la sala, como son el proceso, instalaciones, equipos, flora habitual, flujos de aire, etc.
Adicionalmente, se realizará una monitorización fuera de las operaciones del área, tanto previo a la desinfección como posteriormente, antes y después de la fabricación y después de un periodo de inactividad. El objetivo de esta monitorización será la detección de potenciales incidentes dentro de la zona crítica. Como novedad en la nueva versión del Anexo 1, es necesario plantear la monitorización no solo en las salas críticas de grado A y B sino que se deben establecer planes de monitorización para las salas de grado C y D.
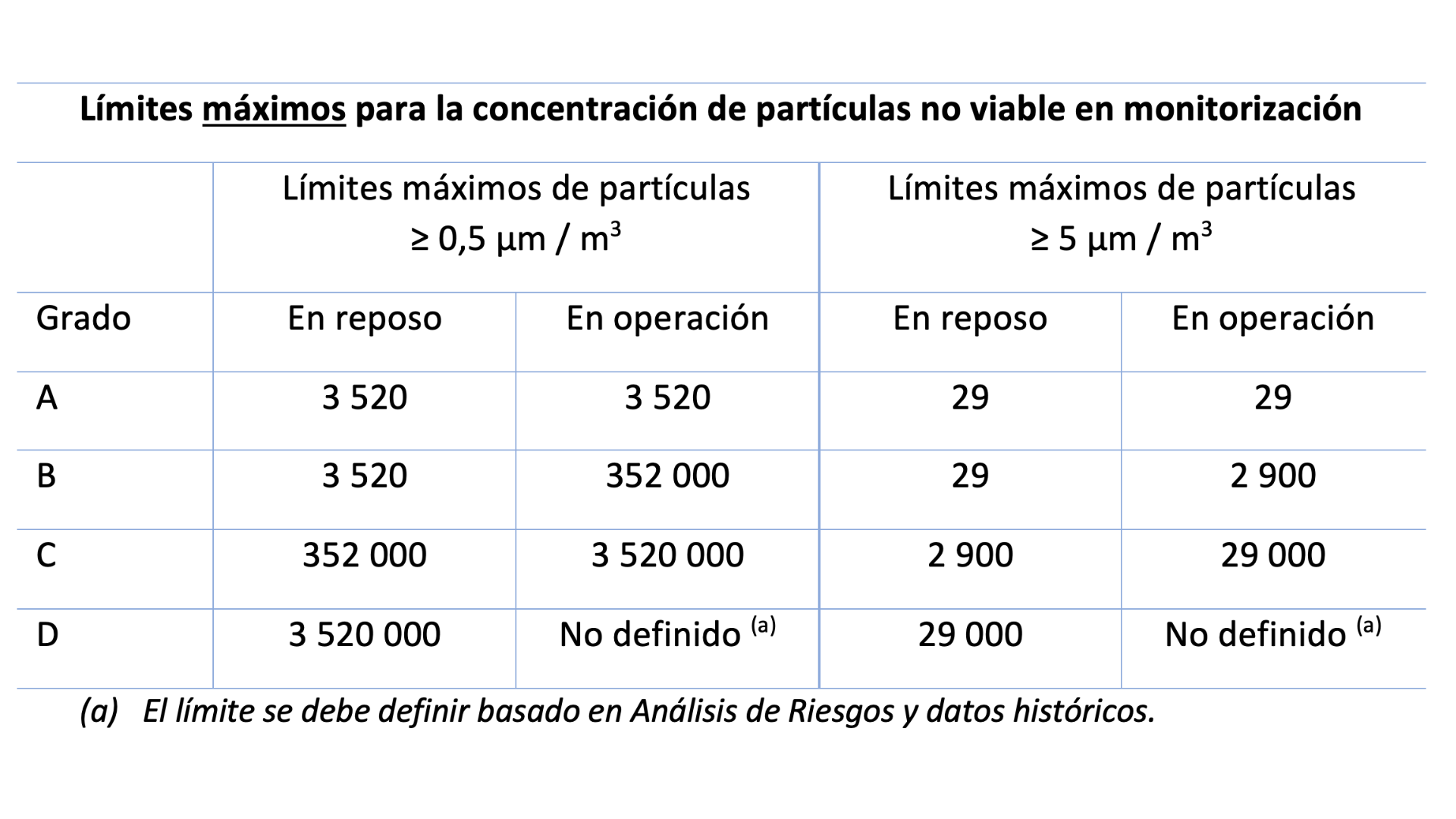
En cuanto a la monitorización de partículas no viables hay un cambio significativo relacionado con las salas Grado D, cuyo límite máximo de partículas en operación ahora se debe fijar de acuerdo a un análisis de riesgos y en datos históricos. Para todas las clases de sala, los límites de alerta se establecerán en función de un análisis de tendencias.
El nuevo borrador propone que la monitorización en continuo de Grado A se haga con un flujo de aire adecuado, que nos indica debe ser de al menos 28 litros por minuto (1 pie cúbico por minuto) lo cual implica la sustitución para aquellos laboratorios que tengan contadores de 2,8 L (0,1 pie cúbico).
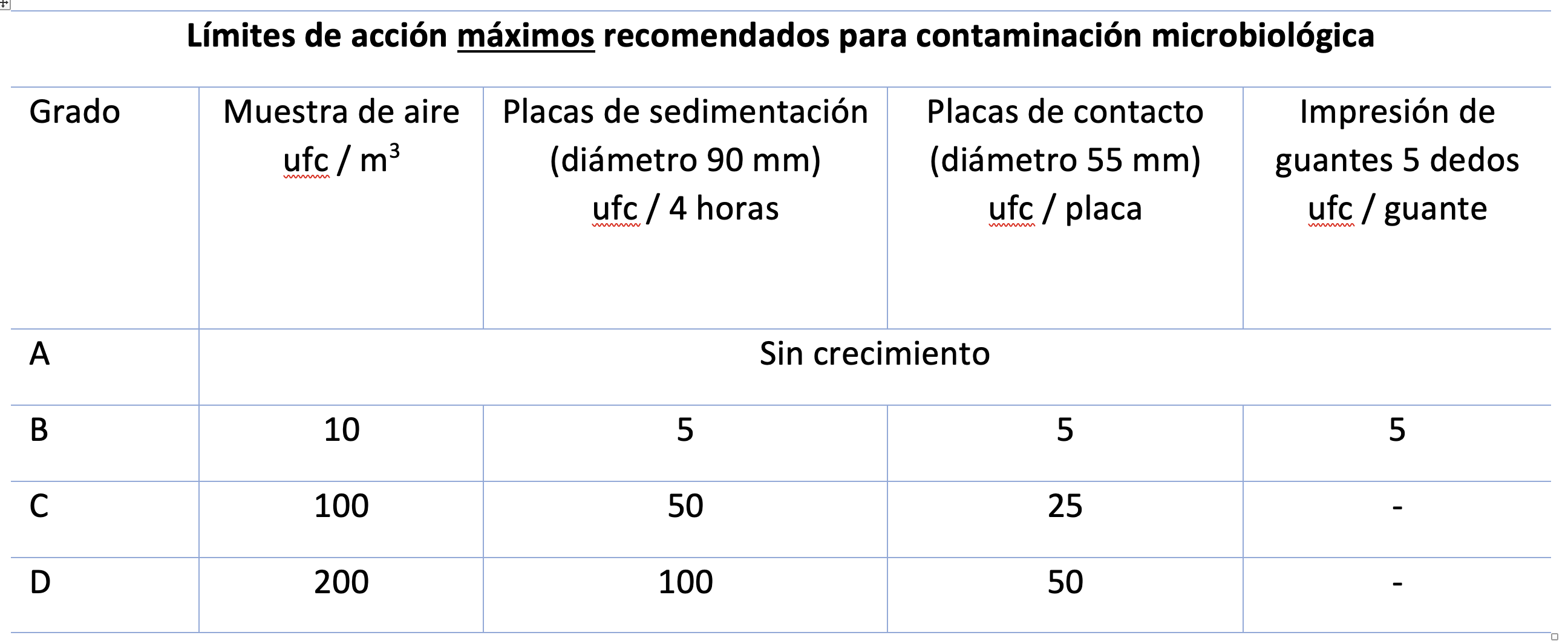
Relativo a la contaminación microbiológica, el cambio más significativo en cuanto a límites es que en Grado A no se permite ningún crecimiento durante la monitorización; hasta ahora existía el resquicio del límite “menor a 1” que servía de justificación en muchos casos para evitar realizar la investigación pertinente. Ahora se especifica en la norma, que cualquier crecimiento debe conllevar una investigación. Adicionalmente se nos especifica que los microorganismos detectados en Grados A y B deben ser identificados hasta el nivel de especie (evaluando además su potencial impacto en los lotes afectados).
Simulación del Proceso Aséptico (APS)
Relativo a la simulación del proceso aséptico hay varios cambios. Por supuesto, el más evidente es el cambio de nombre. Dejamos atrás el término “Media fill” para incorporar el de “Simulación del Proceso Aséptico”.
Otro de los cambios principales es que ya no es una forma de validación de proceso, si no una vía más para monitorizarlo; forma parte del diseño de una estrategia de control de la contaminación del área. Sin embargo, se especifica que debe ser parte de la validación inicial o tras modificaciones significativas de operativa, instalaciones, servicios o equipos. Igualmente se habla de APS como revalidación periódica, por lo que da la impresión de que sigue a caballo entre validación y monitorización.
Se nos dan muchas más pautas para la ejecución de esta prueba, dando detalles que hasta ahora teníamos que buscar en normativas y guías alternativas, con indicaciones para actividades como las intervenciones a incluir, situaciones peor caso, la liofilización, o la incubación de unidades. Aun así, todos los parámetros que se definan en la simulación estarán basados en herramientas de Gestión de Riesgos de la calidad.
Sin embargo, el cambio más relevante y con mayor impacto en relación a la simulación del proceso aséptico está en el criterio de aceptación. Si hasta ahora teníamos un margen de una unidad contaminada en aquellas simulaciones de más de 5.000 unidades, ahora el objetivo es directamente 0 unidades contaminadas; la contaminación de una única unidad supone una simulación fallida y requerirá, por tanto, realizar investigación de la que obtengamos las causas raíces más probables, con la consiguiente implementación de acciones correctivas y la realización de las simulaciones necesarias para demostrar que el proceso vuelve a estar bajo control (aunque deja abierto el número de repeticiones, siempre en relación con herramientas de gestión de riesgos, sí nos indica que normalmente será un mínimo de tres en la validación inicial).
Además de lo anterior, se deberá revisar la documentación de los lotes fabricados en la línea tras la última simulación conforme, para evaluar posibles fallos de esterilidad en los mismos. Aquellos lotes que no hayan sido liberados aún deben quedar en cuarentena, incluyendo los fabricados tras la simulación fallida. La decisión sobre la liberación al mercado de dichos lotes se deberá tomar teniendo en cuenta los resultados de la investigación realizada.
Respecto a la simulación fallida se incluye una frase que podría tener consecuencias económicas para la empresa: “La producción solo debe reanudarse tras completar una revalidación exitosa”. ¿Quiere esto decir que no se puede fabricar a riesgo? ¿Y tras una lectura intermedia de unidades a los 7 días de incubación? La frase parece lo bastante contundente como para justificar lo contrario.

De forma general, en esta revisión del Anexo 1 los controles en personal y monitorización (incluyendo APS) están más racionalizados, quedan relacionados directamente a la calidad de los productos, y por tanto a la seguridad del paciente. Se integra en las operaciones esa visión holística en que se asume que todo lo que se haga tiene impacto en otros elementos del proceso.
¡Seguid atentos a los siguientes artículos en los que seguiremos analizando los cambios en el último borrador del Anexo 1 y sus implicaciones para la industria!
